-
 Être rappelé
Être rappelé
-
Trouver un centre
-
Demander un rendez-vous



 Test acouphènes
Test acouphènes
 Test auditif
Test auditif
Être rappelé
Demandez un rendez-vous
"*" indicates required fields
Être rappelé
Syndrome de Usher : restauration de l’audition et de l’équilibre grâce à la thérapie génique
17.10.2022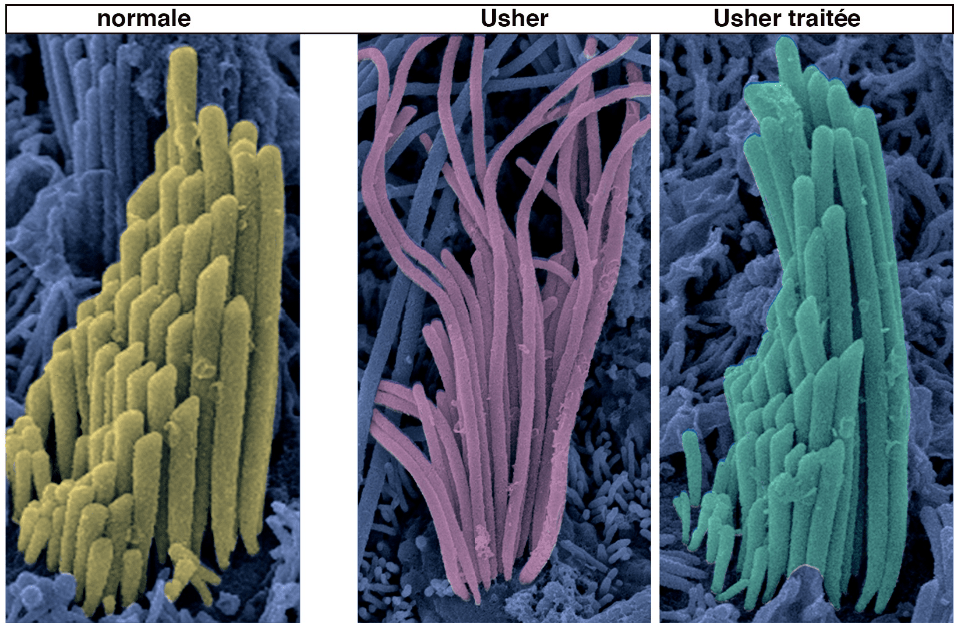
Vers un traitement du Syndrome de Usher
Des chercheurs de l’Institut Pasteur, de l’Inserm et du CNRS ont réussi à rétablir l’audition et l’équilibre dans un modèle murin du syndrome de Usher de type 1G (USH1G) grâce à une thérapie génique. Le gène USH1G, essentiel pour la formation et le maintien de l’appareil de transduction mécano-électrique des cellules sensorielles de l’oreille interne, a été localement injecté, permettant ainsi de restaurer le fonctionnement de cette structure. Ce succès a permis à un modèle murin de ce syndrome de récupérer partiellement l’ouïe et complètement l’équilibre. Les résultats de cette étude, publiée dans la revue PNAS, ouvrent de nouvelles perspectives pour le traitement des formes génétiques de surdité par thérapie génique.
Une surdité liée à un dysfonctionnement de l’oreille interne
La surdité est un déficit sensoriel prévalent dans le monde, touchant plus de 280 millions de personnes selon l’OMS. En France, 1 enfant sur 700 naît avec une surdité sévère ou profonde, et 1 enfant sur 1000 devient malentendant avant l’âge adulte. Au cours des deux dernières décennies, d’importants progrès ont été réalisés dans la compréhension des surdités héréditaires, dont la plupart sont attribuables à un dysfonctionnement de l’oreille interne, composée de l’organe de l’audition (cochlée) et des cinq organes de l’équilibration (saccule, utricule et trois canaux semi-circulaires), abritant les cellules sensorielles ou cellules ciliées. À ce jour, près de 70 gènes responsables de ces surdités ont été identifiés.
Syndrome de Usher et surdité congénitale
Le syndrome de Usher de type 1 (USH1) se caractérise par une surdité congénitale profonde, des troubles de l’équilibration et une atteinte visuelle progressive conduisant à la cécité. Ce syndrome peut résulter de mutations dans 5 gènes différents, dont le gène USH1G, codant pour une protéine cruciale pour la cohésion de la touffe ciliaire des cellules ciliée
Actuellement, les individus atteints de surdité et de troubles de l’équilibre sont généralement équipés de prothèses auditives et peuvent bénéficier d’une rééducation pour améliorer leurs problèmes d’équilibre, mais les résultats varient. Une alternative prometteuse pour traiter les surdités d’origine génétique est la thérapie génique, qui implique le transfert d’une copie saine du gène défectueux pour restaurer l’expression de la protéine manquante. Cependant, jusqu’à présent, seules des améliorations partielles de l’audition avaient été obtenues dans des modèles murins de certaines formes de surdité humaine qui ne présentaient pas de graves anomalies de la structure des cellules ciliées.
Une étude menée par le CNRS visant à restaurer l’audition grâce à la thérapie génique
Dans cette étude, les chercheurs ont restauré l’audition et l’équilibre chez un modèle murin du syndrome USH1 en injectant localement le gène USH1G à l’aide du virus AAV8, sans danger pour la santé mais ciblant spécifiquement les cellules ciliées de l’oreille interne. L’expression du gène médicament a été détectée dès 48 heures après l’injection, montrant ainsi que l’injection unique suffit à améliorer l’audition et l’équilibre chez les souriceaux affectés. Ces résultats suggèrent que la protéine médicament a pu interagir normalement avec les autres protéines du complexe moléculaire USH1, telles que les protéines cadhérine 23, protocadhérine 15, myosine VIIa et harmonine, nécessaires au bon fonctionnement des canaux de la transduction mécano-électrique.
Saaïd Safieddine, directeur de recherche du CNRS à l’Institut Pasteur et dernier auteur de l’étude, a souligné l’importance de cette découverte en déclarant que “nous venons de prouver qu’il est possible de corriger partiellement une forme génétique particulière de surdité accompagnée de troubles de l’équilibre grâce à une thérapie génique locale, effectuée après le stade de développement de l’oreille initialement touché par la mutation responsable. La fenêtre de temps pour traiter efficacement le syndrome USH1 par thérapie génique pourrait donc être plus large qu’initialement envisagée.”
Cette avancée représente une étape cruciale vers la réalisation d’essais cliniques de thérapie génique en vue d’un traitement curatif de certaines formes génétiques de surdité chez l’Homme.